Nowadays, complex communities of microorganisms can be studied in depth with metagenomics and metatranscriptomics driven by evolution of sequencing techniques. Indeed, these meta’omics techniques offer insight concerning structure and functions of the studied communities. Notwithstanding, raw microbiota sequencing data are difficult to analyze due to their size and the numerous available tools [1,2] for analytical workflow. Indeed, sequences analysis requests successive bioinformatics tasks (e.g. quality control, sequence sorting, taxonomic analysis, functional analysis, statistical analysis). Choosing the best tools with correct parameter values and combining tools together in an analysis chain is a complex and error-prone process. Therefore, there is an urgent need for modular, accessible and sharable user-friendly tools.
Several solutions are proposed in the form of bioinformatic pipelines to exploit metagenomic data. Tools such as QIIME [3], Mothur [4], MEGAN [5], IMG/M [6,7] or CloVR-metagenomics [8] are useful tools, but they do not provide a complete analytical workflow or are limited to amplicon sequences. Other interesting solutions such as SmashCommunity [9], RAMMCAP [10], MetAMOS [11] propose complete analysis pipelines. However, these command-line tools without proper graphical interface are not user-friendly solutions for researchers with low programming or informatics expertise. For their part, web-based services (MG-RAST [12,13], EBI metagenomics [14]) analyze microbiota data with automated pipelines working as a black box that also prevents the mastering and monitoring of the data process. Reproducibility can be questioned by this lack of transparency and flexibility. Alternative approaches to improve usability, transparency and modularity can be found in open-source workflow systems (Taverna [15], Wings [16], Galaxy [17,18] or Kepler [19]). They can be used to build a complete modular, accessible, sharable and user-friendly framework to analyze sequence data using chosen tools and parameters.
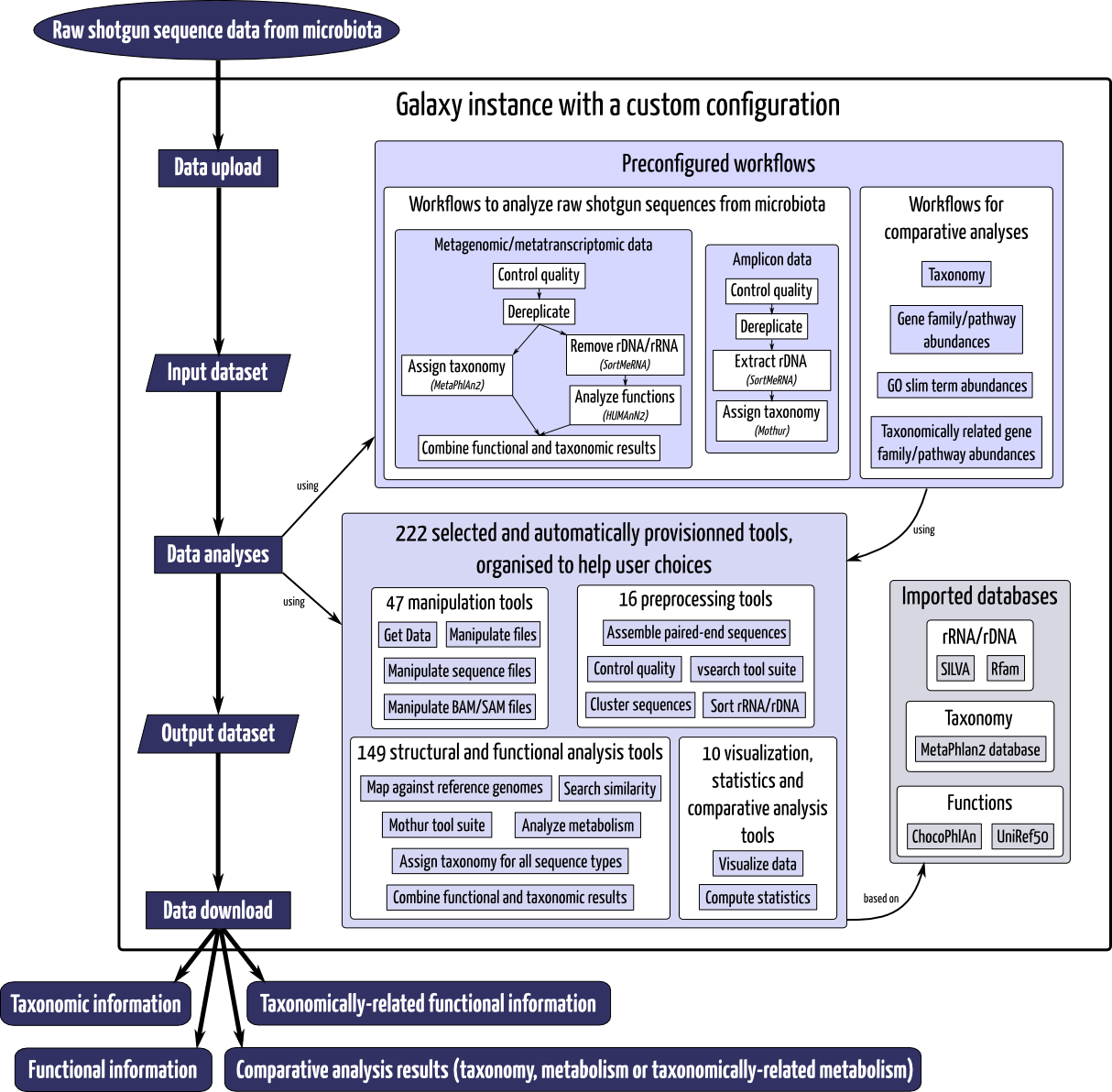
To overcome these limitations, we have developed ASaiM, an open-source opinionated Galaxy-based framework. It is accessible via a web-based interface to ease microbiota sequence data analyses for biologists. With a custom collection of tools, workflows and databases, the framework is dedicated to microbiota sequence analyses (particularly extraction of taxonomic and metabolic information from raw sequences). By its intrinsic modularity, ASaiM allows adjustment of workflows, tools parameters and used databases for general or specific analyses and provides therefore a powerful framework to easily and rapidly analyze microbiota data in a reproducible and transparent environment.

Implementation
Based on a custom Galaxy instance, ASaiM framework is integrating tools, specifically chosen for metagenomic and metatranscriptomic studies and hierarchically organized to orient user choice toward the best tool for a given task. We integrate tools such as FastQ-Join [20] for paired-end assembly, FastQC [21] and PRINSEQ [22] to control quality, vsearch [23] to dereplicate, CD-HIT [24,25] to cluster sequences, SortMeRNA [26] to sort rDNA or rRNA sequences, NCBI Blast + [27,28] and Diamond [29] for similarity search, MetaPhlAn2 [30] to assign taxonomy of sequence type, HUMAnN2 [31] to analyze metabolism, Group HUMAnN2 to GO slim term [32] to get a broad overview of metabolism functions, GraPhlAn, KRONA [33] and R scripts to get graphical outputs and statistical tests. All available tools, their versions are described in ASaiM documentation. These tools can be used alone or orchestrated inside workflows.
To guide users and then ease exploitation of raw metagenomic or metatranscriptomic sequences, we propose a standard but customizable workflow. This workflow is composed of the following steps (i) quality control, (ii) rRNA/rDNA sequence sorting, (iii) taxonomic assignation, (iv) functional assignation to UniRef50 gene families, MetaCyc pathways and GO slim term, and (v) linking of taxonomic and functional analyses. Graphical outputs are generated at the different stages of the workflow (with custom scripts or using dedicated tools). All choices (tools, version, parameters, execution order) are described in ASaiM documentation. Other workflows are also available for comparative analyses of taxonomic and/or functional results obtained with the previously described worklow.
ASaiM framework source code is available under Apache 2 license on GitHub. The code source includes customization of the Galaxy instance: automatic configuration, launch and provision with chosen tools, workflows and needed databases.
Validation
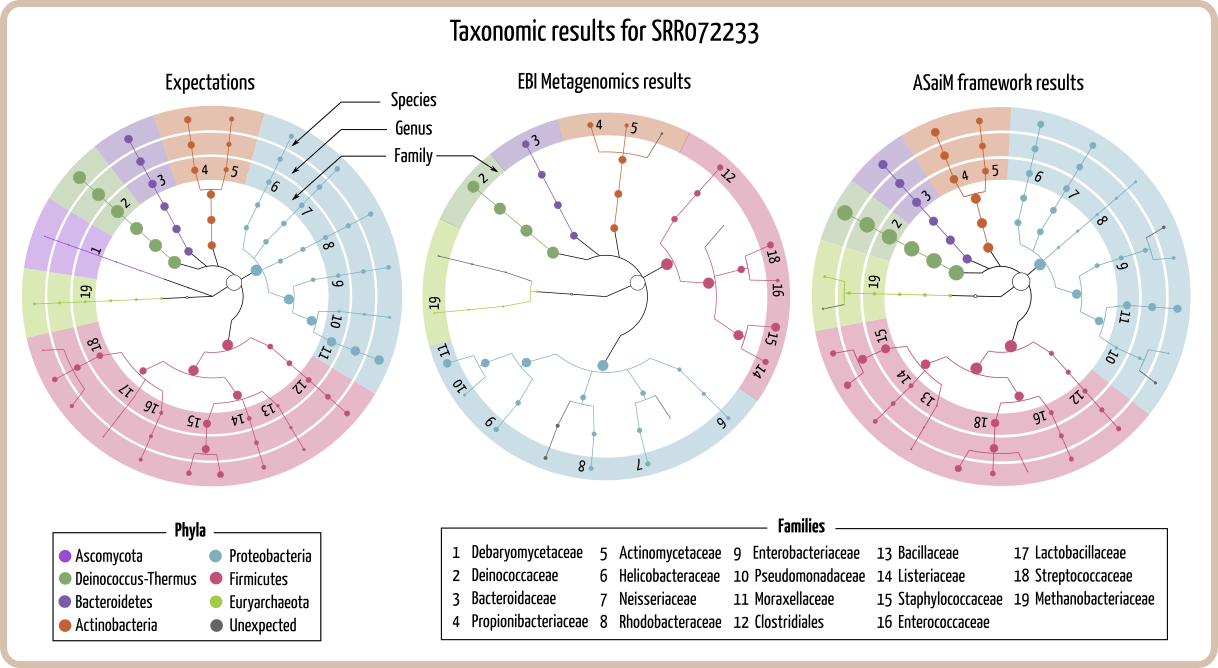
ASaiM framework was tested on two mock metagenomic datasets. These datasets are metagenomic shotgun sequences (>1,200,000 454 GS FLX Titanium single-end sequences) from a controlled microbiota community (with 22 known microbial species), available on EBI metagenomic database. Taxonomic and functional results from ASaiM workflow were compared to the ones obtained with EBI metagenomics pipeline [14] (version 1.0). Details about these analyses (scripts, results, extended report, input data) are available on a dedicated GitHub repository.
For these tests, an ASaiM customed Galaxy instance was deployed on a Debian GNU/Linux System with 8 cores Intel(R) Xeon(R) at 2.40 GHz and 32 Go of RAM. On both datasets, execution of whole main workflow (from quality control to taxonomic and functional analyses integration) lasts less than 5h30, uses maximum of 1.52 Go of RAM.
In both EBI metagenomic pipeline and ASaiM main workflow, raw sequences are pre-processed with first a quality control to remove low quality, small and duplicated sequences and then a rRNA/rDNA sorting. After quality control, more sequences are conserved (>96 %) with ASaiM workflow than with EBI metagenomics pipeline (<87%), despite identical input sequences. The different tools (PRINSEQ [22] and vsearch [23] for ASaiM workflow, Trimmomatic [34], UCLUST [35] and RepeatMasker [36] for EBI metagenomics pipeline) and parameters in EBI and ASaiM workflows may explain the observed differences. The quality-controlled sequences are afterwards sorted in rRNA or non rRNA sequences (with SortMeRNA [26] and rRNASelector [37] for respectively ASaiM and EBI metagenomics ). With both pipelines, the proportion of rRNA sequences is low (<1.5%) as expected for any metagenomic datasets.
These rRNA sequences ([3]. ASaiM workflow uses MetaPhlAn2 [30] on all quality controlled sequences (>1,100,000 sequences), leading to more accurate and statically supported taxonomic assignations, which are also more precise (until species levels, against family levels with EBI metagenomics pipeline). Contrary to taxonomic results from EBI metagenomics pipelines, all genera found with ASaiM workflow correspond to expected genera. Despite previous differences, relative abundances of found all families are similar for both pipelines and are correlated with expected abundances of these mock datasets.
For functional analyses, ASaiM workflow uses HUMAnN2 [31] to extract relative abundance of gene families (UniRef50) and metabolic pathways (MetaCyc). In EBI metagenomics pipeline, functional analyses are only based on InterPro similarity search. In both pipelines, gene families or proteins are afterwards grouped into slim Gene Ontology term (cellular components, biological processes and molecular functions), with relative abundance. In ASaiM, this step based on UniRef50 gene families is performed by specially developed tool [32]. The variability of relative abundances of GO slim terms between both pipelines explains at most 35% of overall variability of relative abundances of GO slim terms. Therefore, GO slim term differences between both pipelines exist, but they are negligible.
In addition, in HUMAnN2 results, abundances of gene families and pathways are stratified at community level. The taxonomic information (abundance of given pathway or gene families for a given taxon) can be related to global taxonomic information and, particularly, species abundances, in ASaiM workflow. This type of information is not available with EBI metagenomic pipeline. A strong correlation is observed between gene family or pathway mean abundances and related species abundances inside both datasets.
Conclusion
With an easy-to-install custom Galaxy instance, ASaiM provides a simple and intuitive preconfigured environment to study microbiota. The build workflow with chosen tools, parameters and databases allows a rapid (few hours on a standard computer), easy, reproducible, complete and accurate analysis of a metagenomic or metatranscriptomic dataset from raw sequences to taxonomic and functional analysis. This workflow as well as workflows for comparative analyses are proposed, but in each case their tools with their parameters and execution order are customizable by the user. To help in tool, parameter and workflow choices, a documentation is available on ReadTheDocs. ASaiM framework is then a powerful framework to analyze shotgun raw sequence data from complex communities of microorganisms. This open-source and biologist-oriented solution enhances usability, reproducibility and transparency of such studies.
References
[1] M.L.Z. Mendoza et al, Brief Bioinform 16 (2015) 745–758.
[2] N. Segata et al, Molecular Systems Biology 9 (2013) 666.
[3] J.G. Caporaso et al, Nature Methods 7 (2010) 335–336.
[4] P.D. Schloss et al, Appl. Environ. Microbiol. 75 (2009) 7537–7541.
[5] D.H. Huson et al, Genome Res. 17 (2007) 377–386.
[6] V.M. Markowitz et al, Nucl. Acids Res. 42 (2014) D568–D573.
[7] V.M. Markowitz et al, Nucl. Acids Res. 36 (2008) D534–D538.
[8] S.V. Angiuoli et al, BMC Bioinformatics 12 (2011) 356.
[9] M. Arumugam et al, Bioinformatics 26 (2010) 2977–2978.
[10] W. Li, BMC Bioinformatics 10 (2009) 359.
[11] T.J. Treangen et al, Genome Biol. 14 (2013) R2.
[12] R.K. Aziz et al, BMC Genomics 9 (2008) 75.
[13] F. Meyer et al, BMC Bioinformatics 9 (2008) 386.
[14] S. Hunter et al, Nucl. Acids Res. 42 (2014) D600–D606.
[15] K. Wolstencroft et al, Nucl. Acids Res. 41 (2013) W557–W561.
[16] Y. Gi et al, IEEE Intelligent Systems 26 (2011) 62–72.
[17] D. Blankenberg et al, Curr Protoc Mol Biol 0 19 (2010) Unit–19.1021.
[18] J. Goecks et al, Genome Biology 11 (2010) R86.
[19] B. Ludäscher et al, Concurrency Computat.: Pract. Exper. 18 (2006) 1039–1065.
[20] E. Aronesty, Ea-Utils : “Command-Line Tools for Processing Biological Sequencing Data,” 2011.
[21] S. Andrews, FastQC: A Quality Control Tool for High Throughput Sequence Data, 2010.
[22] R. Schmieder, R. Edwards, Bioinformatics 27 (2011) 863–864.
[23] T. Rogne et al, Vsearch: VSEARCH 1.4.0, 2015.
[24] L. Fu et al, Bioinformatics 28 (2012) 3150–3152.
[25] W. Li, A. Godzik, Bioinformatics 22 (2006) 1658–1659.
[26] E. Kopylova et al, Bioinformatics 28 (2012) 3211–3217.
[27] P.J. Cock et al, GigaScience 4 (2015) 39.
[28] C. Camacho et al, BMC Bioinformatics 10 (2009) 421.
[29] B. Buchfink et al, Nat Meth 12 (2015) 59–60.
[30] D.T. Truong et al, Nat Meth 12 (2015) 902–903.
[31] S. Abubucker et al, PLoS Comput Biol 8 (2012) e1002358.
[32] B. Batut, group_humann2_uniref_abundances_to_GO: First Stable Version, 2016.
[33] B.D. Ondov et al, BMC Bioinformatics 12 (2011) 385.
[34] A.M. Bolger et al, Bioinformatics 30 (2014) 2114–2120.
[35] R.C. Edgar, Bioinformatics 26 (2010) 2460–2461.
[36] A. Smit et al, RepeatMasker Open-3.0, 1996.
[37] J.-H. Lee et al, J Microbiol. 49 (2011) 689–691.